Transcriptome sequencing (RNA-Seq) produces big amounts of valuable information on all transcribed elements in the genome. With RNA-Seq, researchers can interrogate the transcriptome to profile gene expression, uncover alternative splicing, and identify novel and discard aberrant transcripts and coding variants, among others. The resulting BAM Files are the source of information for all proceeding step
Why is BAM File Quality Control important?
To make RNA-seq data more reliable, scientists have to overcome biases and limitations intrinsic to RNA-Seq protocols, such as nucleotide composition bias, GC bias, PCR bias, etc. Additionally, specific RNA-Seq quality control metrics, including sequencing depth, read distribution coverage, and coverage uniformity are even more important. These metrics assure that the experiment provides sufficient read information and that it is evenly distributed across the transcriptome. Consequently, sequencing depth must be saturated before carrying out many RNA-Seq applications, including expression profiling, alternative splicing analysis, novel isoform detection, and transcriptome reconstruction. The use of RNA-Seq data with unsaturated sequencing depth results in non-reliable estimations and low abundant splice junctions are not detected. However, sequencing depth is directly proportional to the necessary budget which makes it important to find the right balance.
FastQC, htSeqTool, and FASTX-ToolKit focus entirely on raw sequence-related metrics, RSeQC addresses directly the need of assessing RNA-Seq data comprehensively taking into account the metrics explained above. RSeQC contains basic modules to evaluate raw sequence quality, and RNA-Seq specific modules to perform annotation-based checking of well-annotated organisms (mouse, fly, C. elegans, yeast, etc.). BAM File Quality Control in OmicsBox is based on RSeQC which provides the important metrics and visualizations to fully evaluate RNA-Seq data.
Exemplary Metrics
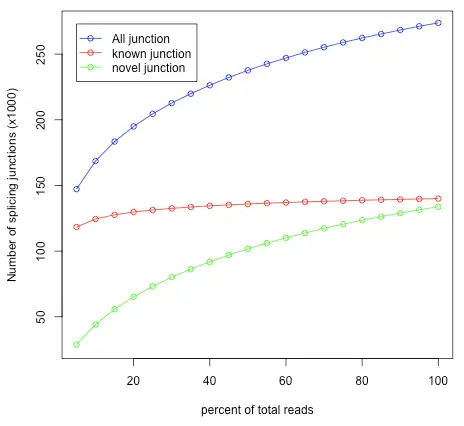
Junction Saturation determines if the current sequencing depth is sufficient to perform alternative splicing analyses. Splice junctions are detected for each re-sampled subset of reads, and the number of detected splice junctions will increase as the resample percentage increases until reaching an asymptotic value. Therefore, the junction saturation test is very important for alternative splicing analysis, as using an unsaturated sequencing depth would miss many rare splice junctions.
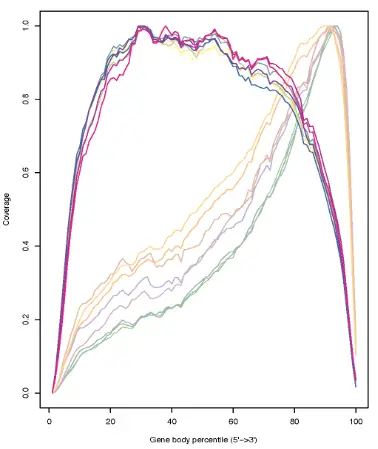
Gene Body Coverage scales all transcripts to 100 nucleotides and calculates the number of reads covering each nucleotide position. Finally, it generates a plot illustrating the coverage profile along the gene body. In the example above we can see that one group of samples covers all gene bodies more uniformly, compared to the other group where the read mapping is strongly biased toward the 3′ end.
BAM File Quality Control in OmicsBox
- BAM-QC can be found in the Transcriptomics module, right below RNA-Seq Alignment. Its metrics have been designed for RNA-Seq data.
- It is possible to provide single and paired-end BAM alignment files. Providing annotations (BED/GFF/GTF) allows for more plots and metrics. The annotations have to match the genome version that was used for the alignment.
- Annotations are accepted in 12-column BED format or GFF/GTF, while the latter will be converted automatically to BED internally.
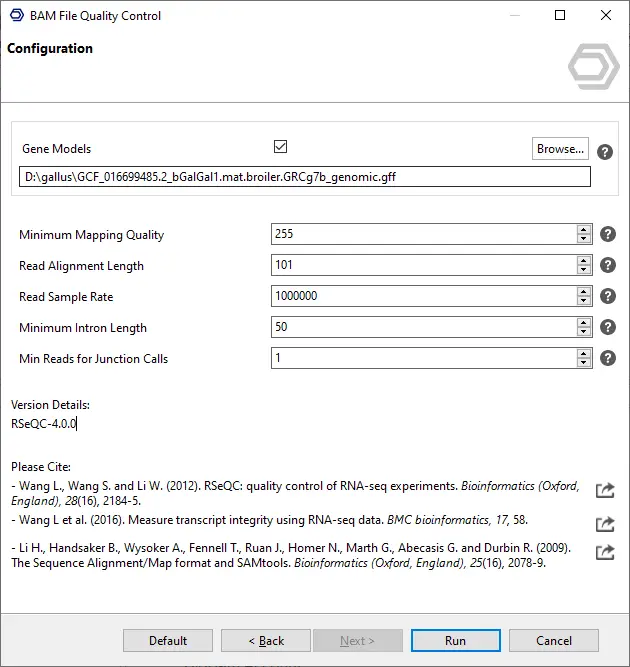
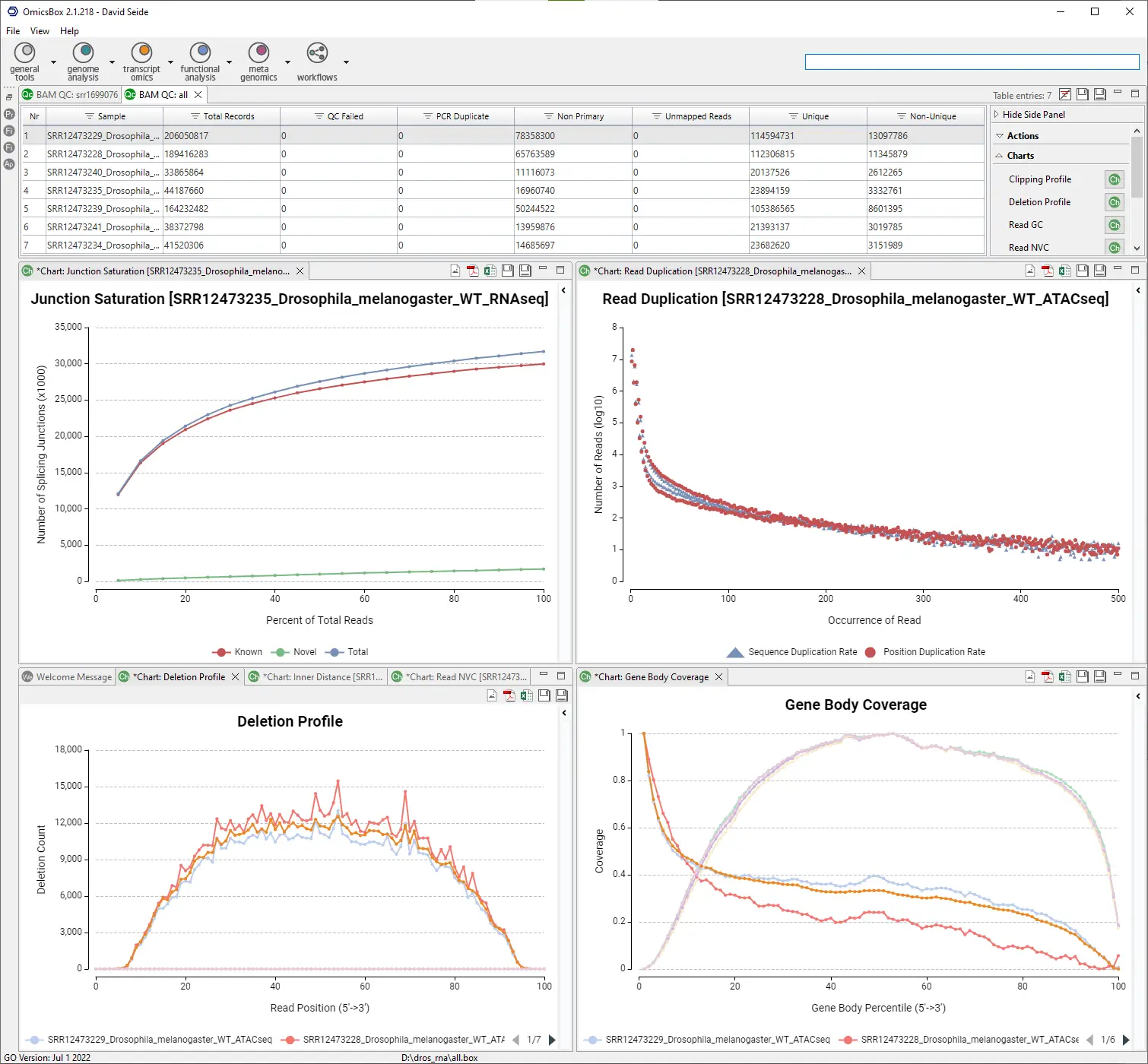
References
- Wang L., Wang S. and Li W. (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics (Oxford, England), 28(16), 2184-5.
- Wang L et al. (2016). Measure transcript integrity using RNA-seq data. BMC Bioinformatics, 17, 58.
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G. and Durbin R. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England), 25(16), 2078-9.
Useful links

About the Author
 David Seide
David Seide is a skilled bioinformatics professional with a BSc in Computer Science and a Master's degree in Bioinformatics. With over 10 years of experience, he currently holds the position of Head of Bioinformatics at BioBam.
In addition to his role, David serves as the Product Owner responsible for the Metagenomics module in OmicsBox, demonstrating his expertise and leadership in the field.
David Seide
David Seide is a skilled bioinformatics professional with a BSc in Computer Science and a Master's degree in Bioinformatics. With over 10 years of experience, he currently holds the position of Head of Bioinformatics at BioBam.
In addition to his role, David serves as the Product Owner responsible for the Metagenomics module in OmicsBox, demonstrating his expertise and leadership in the field.
")
