Long-read sequencing has gained widespread popularity in many areas of biological research due to the many advantages these technologies offer over short reads, as well as the decreasing error rates and costs. Nature Methods named it the method of the year 2022. The field of transcriptomics shows particular interest in long reads, as long-read RNA-sequencing can shed light on the variety of isoforms created through alternative splicing. Multiple isoforms of the same gene can often have different, sometimes even opposite, functional properties, highlighting the importance of conducting analyses at isoform-resolution to uncover the regulation of isoform expression between different conditions of an experiment.
In our quest to make these rapidly evolving technologies more accessible to researchers, OmicsBox has implemented various computational tools for analyzing long-read data and offers a comprehensive suite of end-to-end analysis capabilities.
In this use case, we will explore how OmicsBox can help with the following analysis steps:
- Pre-processing of long-read RNA-sequencing data
- Discovery of novel isoforms & curation of a high-quality transcriptome
- Quantification of short or long reads based on a custom transcriptome
- Functional annotation and pathway analysis at isoform-resolution
Data set
The data set for this use case is based on a publication by Tseng et al. from 2022 titled “Long-read isoform sequencing reveals tissue-specific isoform expression between active and hibernating brown bears (Ursus arctos)”. In this study, the authors examine the differences between active and hibernating brown bears using PacBio long-read RNA-sequencing, in combination with short reads from a previous publication.
This use case will analyze a subset of their publicly available data with the following characteristics:
- 3 brown bears (Ursus arctos horribilis; 2 male, 1 female).
- From each bear, samples of adipose tissue in the summer active and winter hibernation phases, for a total of 6 samples.
- Pacific Biosciences Iso-Seq RNA-Sequencing (via cDNA amplification) on a Sequel system.
Note: while this example uses PacBio data, all steps after the PacBio-specific pre-processing can also process long-read data originating from Oxford Nanopore Technologies‘ platforms.
Data pre-processing
After having downloaded the publicly available reads as Circular Consensus Sequences (CCS) in the ccs.bam format, the first step is to perform pre-processing using the Iso-Seq pipeline. Specifically, this will remove the primers that are still present in the data at this point, and remove any artificial concatemers, which are fusions of 2 cDNA fragments. Although it is not necessary in this data set, this pre-processing pipeline can also segment reads originating from Kinnex technology, as well as demultiplex reads based on barcodes.
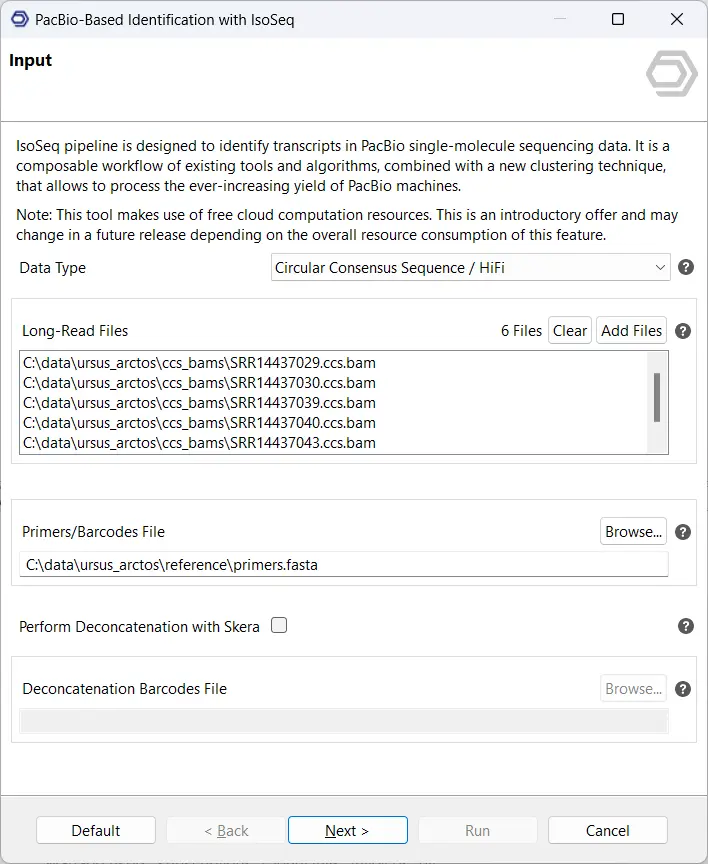
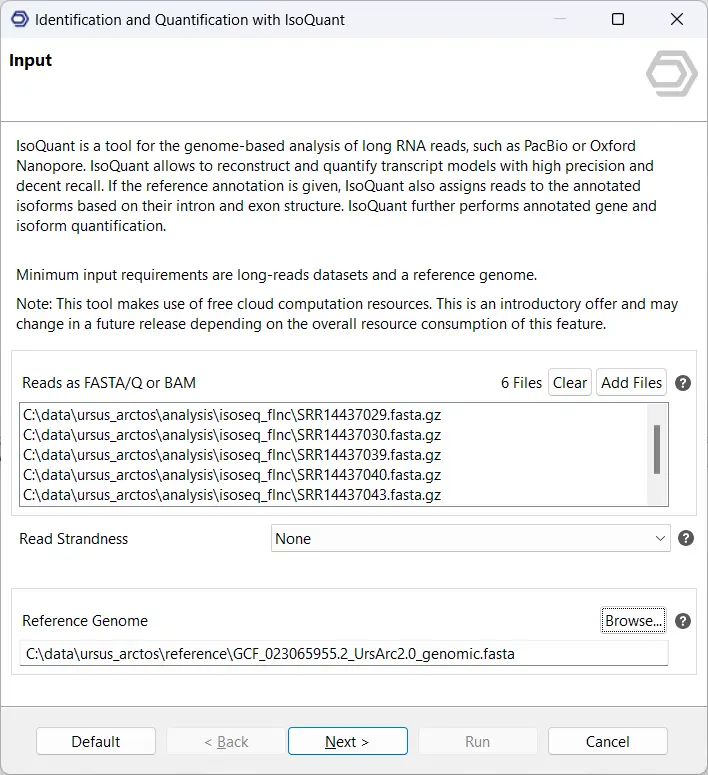
The IsoSeq pre-processing pipeline is located in the Transcriptomics Module, under the sections Long-Read Analysis and Transcript Identification, and PacBio-Based Identification with IsoSeq. As seen in Figure 2, the input consists of 6 samples in the ccs.bam format as well as the relevant primers. This will provide the pre-processed so-called “Full-Length Non-Chimeric” reads, or FLNC reads for short, which we will use for further analysis.
Discovery of novel isoforms
OmicsBox offers many options to create a custom transcriptome, including the discovery of novel isoforms, from long-read data:
- PacBio IsoSeq can be used to create a transcriptome, for which it requires long reads and a reference genome.
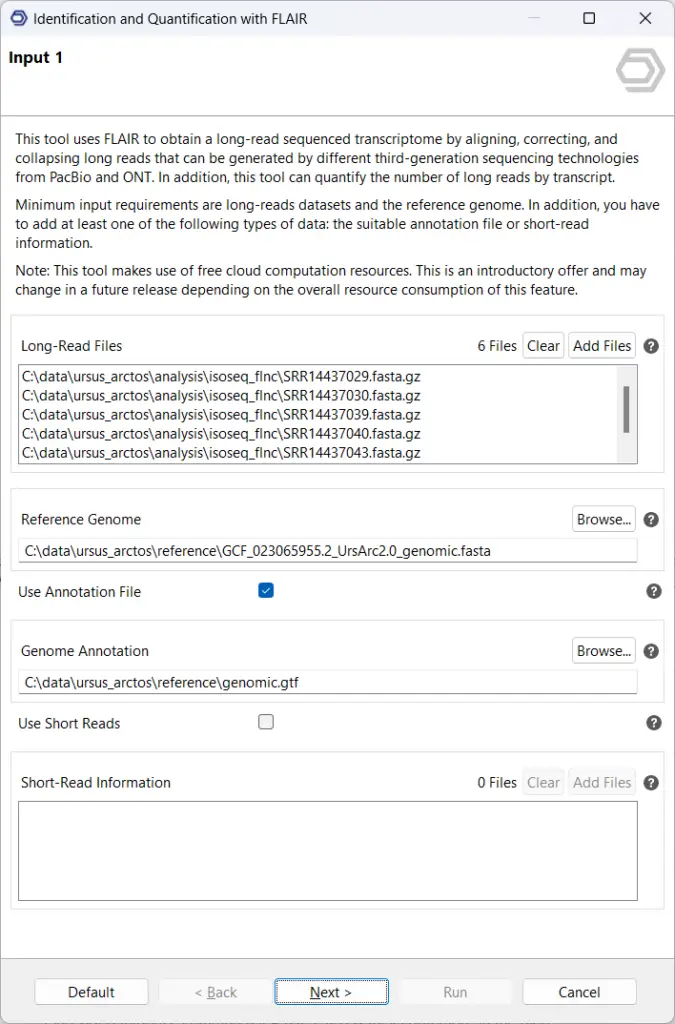
- FLAIR is another option, which requires long reads, a reference genome, and either a reference transcriptome annotation (GTF), or supporting short reads (or both). It works best when supporting short reads are provided, which are used to validate novel splice junctions.
- IsoQuant requires long reads, and a reference genome, and can optionally also use an existing reference annotation (GTF).
- Suppose a transcriptome is to be assembled completely de-novo, e.g. for non-model organisms where a reference genome is not available. In that case, the isONpipeline can be used based only on long reads.
For this use case, we chose to use FLAIR, as the LRGASP challenges recommend it for the study of novel isoforms. This decision has been motivated by this comprehensive benchmarking effort, which has recently been published in Nature Methods and to which BioBam is proud to have contributed.
While the brown bear is not a particularly well-studied organism, there are publicly available reference genomes and transcript annotations, which constitute the input to FLAIR together with the pre-processed long reads. This produces a custom transcriptome that captures a wide variety of novel isoforms.
Curation of a high-quality transcriptome
Once FLAIR has created a transcriptome, it is highly recommended that this transcriptome be further curated to ensure that downstream analysis considers only high-quality, high-confidence isoforms. To take a more detailed look at the transcriptome and filter out any potentially spurious transcript models, OmicsBox offers SQANTI3.
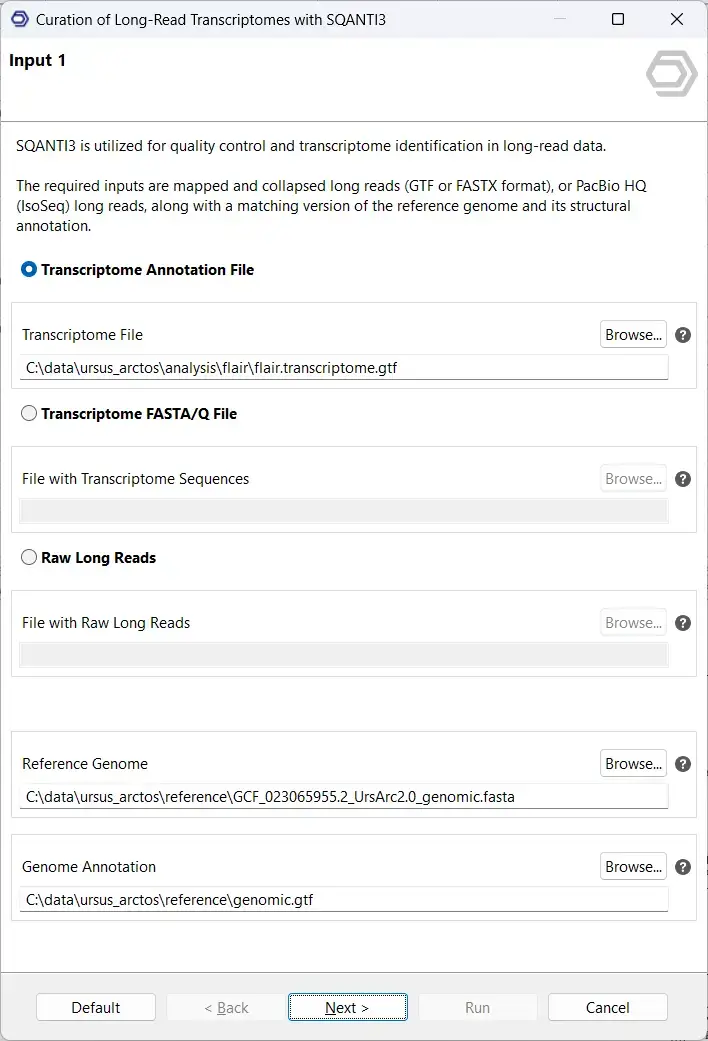
Primarily, SQANTI3 compares the custom transcriptome with a pre-existing reference transcriptome to characterize any novel isoforms. Providing more additional orthogonal data, such as short reads, polyA-motifs, and CAGE-Seq and Quant-seq peaks, will result in a more detailed characterization. We generally recommend providing as many forms of orthogonal data as available
.
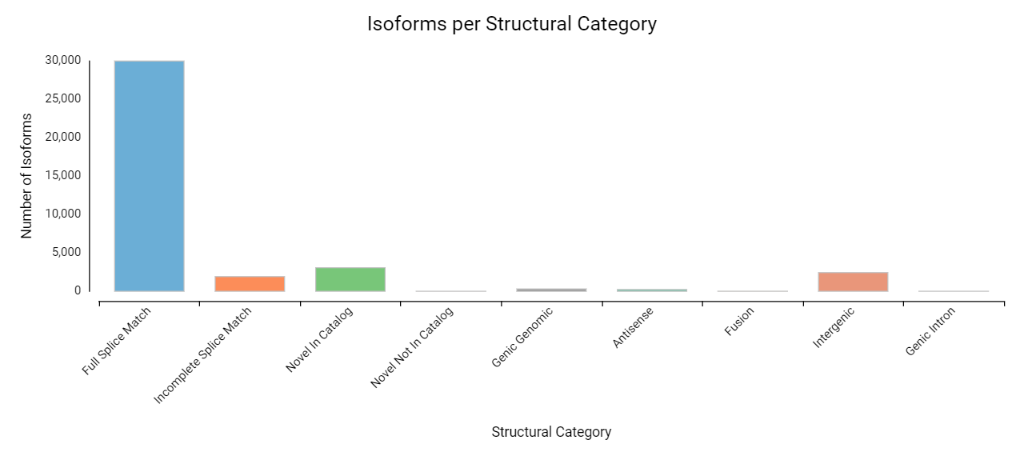

After characterizing the transcriptome, SQANTI3 can also filter out any low-confidence transcripts based either on a set of rules or on a machine learning approach. OmicsBox offers the exploration of these results through a variety of plots, which visualize the characterization of the transcript models into different categories, as well as how many isoforms were filtered out.
Quantification
Since SQANTI3 has curated a high-quality transcriptome, it is necessary to quantify the original set of pre-processed reads based on this refined set of isoforms. It is also possible to quantify short reads based on this transcriptome, for which OmicsBox offers RSEM. However, in this use case, the focus lies on long reads, which is why IsoQuant will perform the quantification based on the long-read data.
However, when quantifying long reads, it is important to keep in mind that this approach comes with some biases. For instance, shorter transcripts may falsely appear to have higher expression than longer transcripts. BioBam is currently contributing to some exciting research together with our collaborators, which will hopefully shed some light on these biases and inform researchers on which analysis strategies can mitigate these problems.
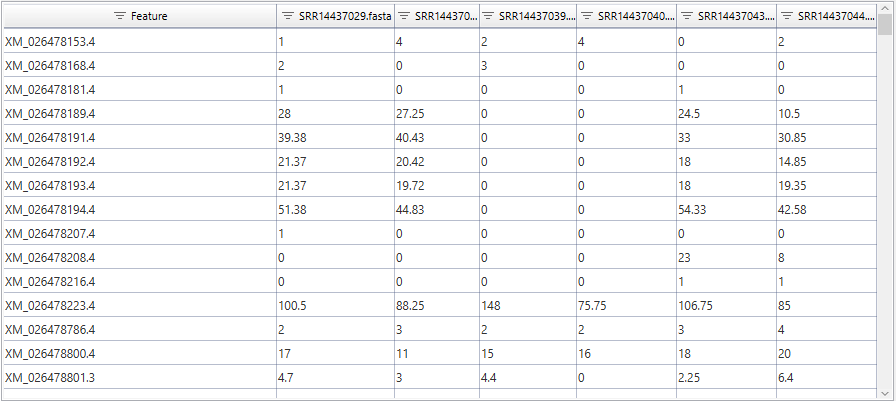
IsoQuant provides the quantification results in the form of two tables (gene- and transcript-level), which are separated into 6 columns for the 6 provided samples. These count tables are ready for further analysis, which will shed light on the observable differences between hibernation and active phases, as well as the functional properties of the sequences.
Differential Expression Analysis
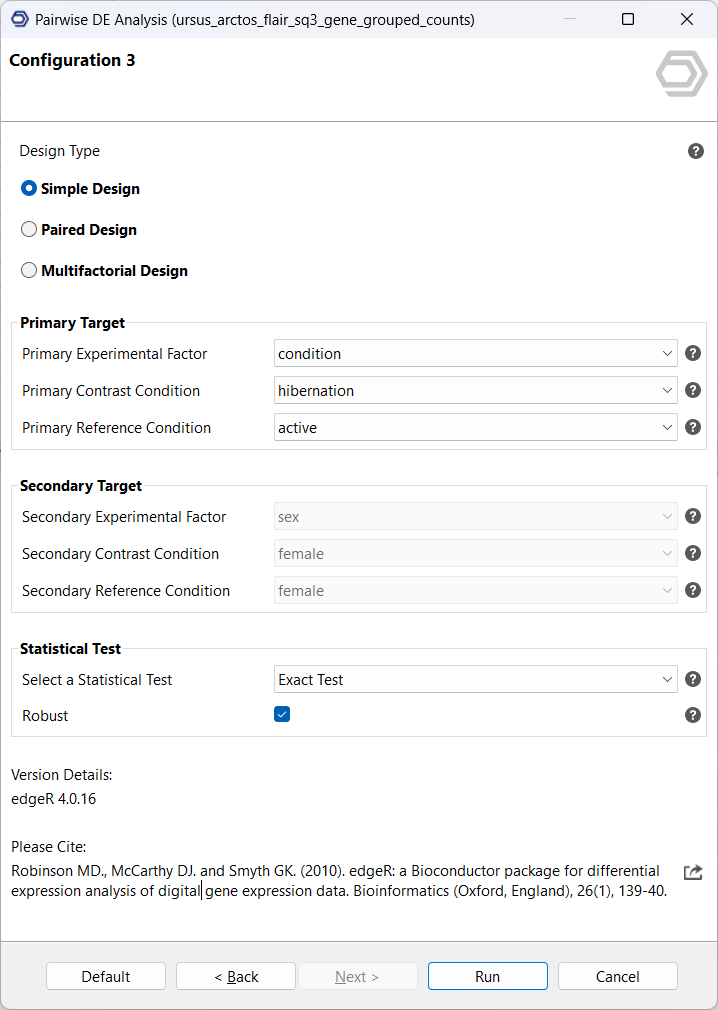
OmicsBox offers a variety of differential expression analysis tools, useful for different experimental designs. For this use case of comparing 2 conditions with 3 samples each, edgeR is the most suitable choice.
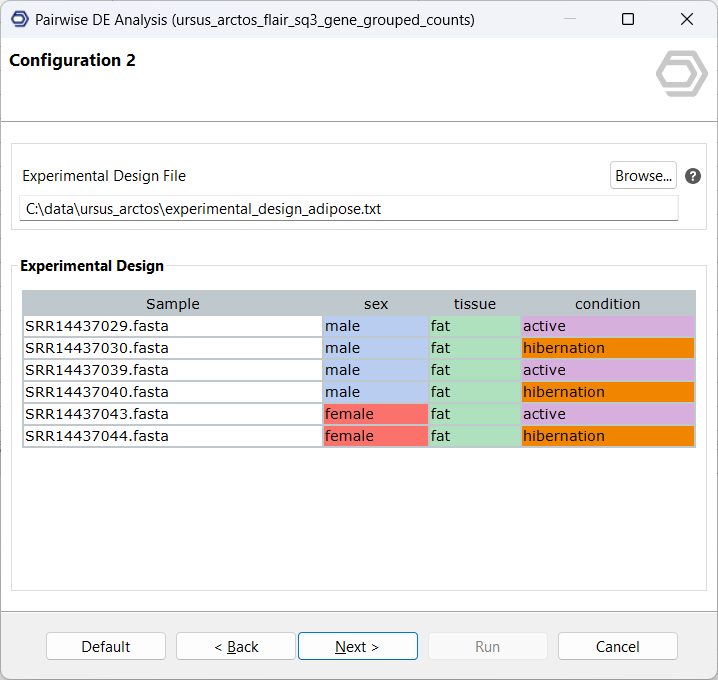
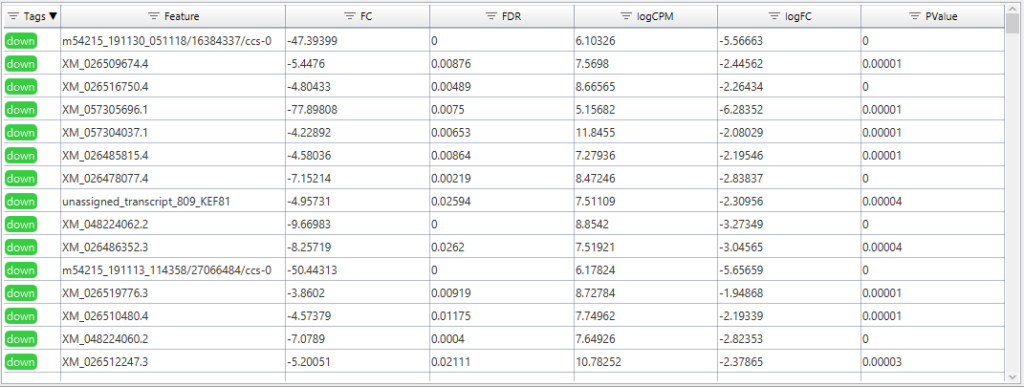
The input consists of the quantification data and a previously prepared text file outlining the experimental design. For this analysis, a simple test design will compare hibernation samples as the contrast, to active samples as the reference. This analysis will be performed twice, once on the gene-, and once on the isoform-level counts.
Pathway Analysis at isoform-resolution
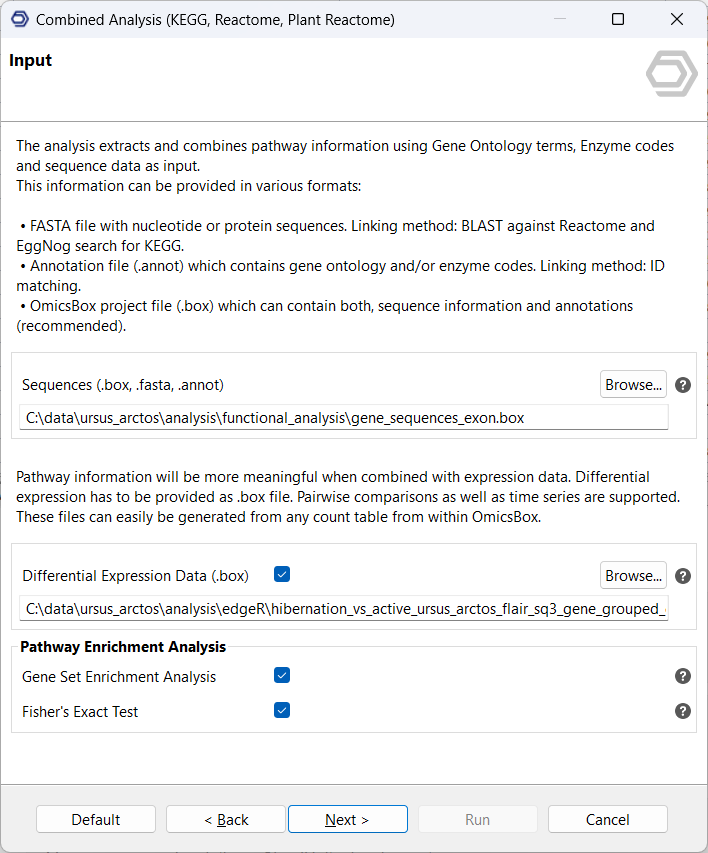
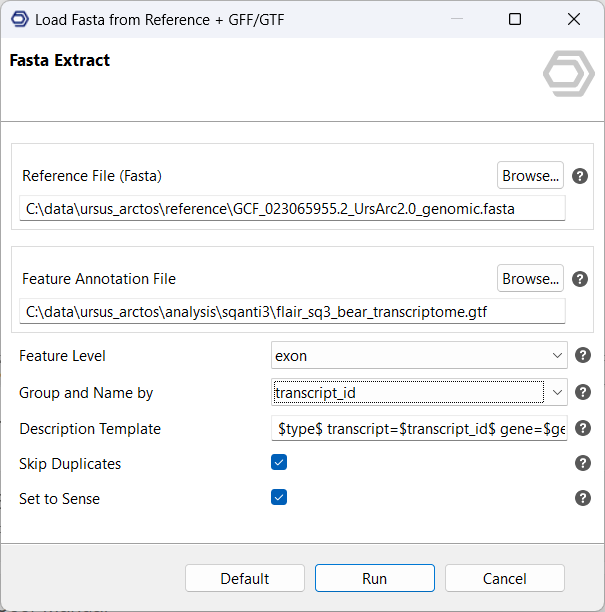
In the final analysis step of this use case, a Combined Pathway Analysis will examine the impact of gene and isoform sequences on a variety of pathways. First, OmicsBox can extract the gene and transcript sequences using a tool located in Functional Analysis, Load, Load Sequences, and Load Fasta from Reference + GFF/GTF. This tool captures exon-sequences from the reference genome based on the transcriptome annotation and, based on whether it groups by gene id or by transcript id, it will provide gene- or transcript-sequences, respectively.
Using this set of sequences, the next step is a Combined Pathway Analysis. This links up the sequences to the KEGG and Reactome (or Gramene for plants) pathway databases using the eggNOG-mapper, as well as the BLAST2GO methodology.
This facilitates the identification of a set of orthologous pathways to which the sequences may be relevant, especially if they are differentially expressed between the two conditions. To investigate the provided results more closely, individual pathway diagrams can be opened and their associated sequences examined in more detail.
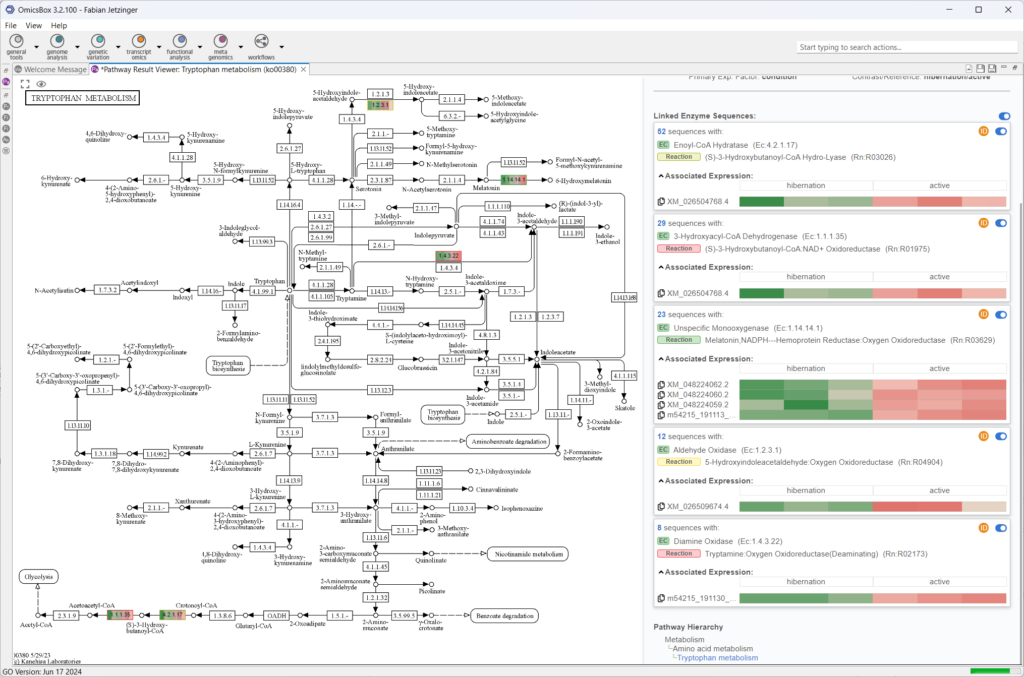
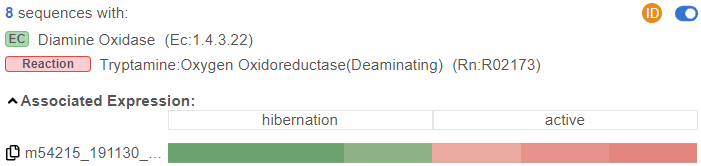
One interesting case in this specific analysis may be the KEGG pathway map for Tryptophan metabolism. While the gene-level analysis already shows several potentially interesting reactions, conducting the same analysis at isoform-resolution yields a much greater level of detail. Interestingly, in one specific reaction, the results display several transcript sequences that are associated with the enzyme code for “Diamine Oxidase”. Out of these transcripts, however, only one is differentially expressed between the two conditions of hibernation and active phases, and, based on the name of the transcript, this is indeed a novel isoform we discovered with FLAIR.
Conclusions
The use case described in this blog post demonstrates how OmicsBox can power in-depth and multi-faceted analyses of long-read data, from pre-processing to functional analysis. Furthermore, we have shown how long reads enable researchers to conduct their transcriptomics analyses at isoform-resolution, creating a more accurate and detailed picture of complex biological processes. This enables the discovery of novel biological insights, which may not have been visible without long-read data.
Specifically, this analysis of long-read data originating from adipose tissue in hibernating and active brown bears has revealed a differentially expressed novel isoform with potential relevance to tryptophan metabolism. Such novel discoveries could contribute to understanding how brown bears facilitate the complex metabolic changes between these two conditions. Without long-read data, such fine-grained differences in complex isoform expression profiles would remain invisible to researchers.
BioBam is committed to bridging the gap between researchers and complex bioinformatic analyses. In addition to continuously updating and expanding OmicsBox, we are also contributing to cutting-edge research to better understand the unique characteristics of long reads.
Sparked your interest? Keep reading our recent blog posts:
- Cutting-edge long-read transcriptomics research published in Nature Methods
- Single Cell RNA-Seq analysis of Arabidopsis thaliana roots
- Population Structure Analysis with OmicsBox
References
Marx, Vivien. “Method of the year: long-read sequencing.” Nature Methods 20.1 (2023): 6-11. https://doi.org/10.1038/s41592-022-01730-w
Pardo-Palacios, Francisco J., et al. “Systematic assessment of long-read RNA-seq methods for transcript identification and quantification.” Nature methods (2024): 1-15. https://doi.org/10.1038/s41592-024-02298-3
Tseng, Elizabeth, et al. “Long-read isoform sequencing reveals tissue-specific isoform expression between active and hibernating brown bears (Ursus arctos).” G3 12.3 (2022): jkab422. https://doi.org/10.1093/g3journal/jkab422
Tang, Alison D., et al. “Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns.” Nature communications 11.1 (2020): 1438. https://doi.org/10.1038/s41467-020-15171-6
Pardo-Palacios, Francisco J., et al. “SQANTI3: curation of long-read transcriptomes for accurate identification of known and novel isoforms.” Nature Methods 21.5 (2024): 793-797. https://doi.org/10.1038/s41592-024-02229-2
Prjibelski, Andrey D., et al. “Accurate isoform discovery with IsoQuant using long reads.” Nature Biotechnology 41.7 (2023): 915-918. https://doi.org/10.1038/s41587-022-01565-y
Chen, Yunshun, et al. “edgeR 4.0: powerful differential analysis of sequencing data with expanded functionality and improved support for small counts and larger datasets.” bioRxiv (2024): 2024-01. https://doi.org/10.1101/2024.01.21.576131
Conesa, Ana, et al. “Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research.” Bioinformatics 21.18 (2005): 3674-3676. https://doi.org/10.1093/bioinformatics/bti610
Cantalapiedra, Carlos P., et al. “eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale.” Molecular biology and evolution 38.12 (2021): 5825-5829. https://doi.org/10.1093/molbev/msab293
About the Author

")
