Constructing a high-quality genome assembly is a complex and challenging process. De novo assemblies often suffer from fragmentation, gaps, and various types of assembly errors, which can compromise downstream analyses. Ensuring the completeness and accuracy of these assemblies is essential for robust biological insights. This is where BUSCO (Benchmarking Universal Single-Copy Orthologs) comes into play.
BUSCO is a widely used tool for evaluating the completeness of genome assemblies, gene annotations, and transcriptomes by assessing the presence of evolutionarily conserved genes. Specifically, it looks for single-copy orthologs that are expected to exist universally across certain taxonomic groups. By focusing on these core genes, BUSCO provides an objective metric to assess how well an assembly captures the expected gene content for a given organism.
The process works by comparing the genome assembly to a curated database of orthologous genes, OrthoDB, that are considered conserved across a broad set of species. BUSCO then classifies these genes into four categories: complete, duplicated, fragmented, or missing, offering a clear picture of the assembly’s quality. This standardized approach makes it easy to not only evaluate genome completeness but also to compare assemblies across different studies or species, facilitating meaningful comparative genomics and guiding improvement of assembly workflows.
How to use BUSCO in OmicsBox
Setting Up BUSCO in OmicsBox
BWA can be found inside the Genome Analysis module, in the “DNA-Seq Assembly” section. The analysis is easily configurable, requiring only the genome assembly, the mode (genome or proteins), the lineage, and the significance threshold (Figures 1 and 2).
A wide range of lineages from the six major phylogenetic clades (Bacteria, Archaea, Eukaryota, Protists, Fungi, and Plants) is available. It is possible to find a complete list of the available lineages in the OmicsBox User Manual.
BUSCO output in OmicsBox
Once the execution finishes, the results will be automatically opened in the platform (Figure 3). The main result is a table (Figure 3-A) containing one row per BUSCO ID for the selected lineage, each colored depending on the given tag:
- COMPLETE: The sequence of the BUSCO ID has been found complete, and a single copy is in the assembly.
- DUPLICATED: the sequence of the BUSCO ID has been found complete but multiple times in the assembly.
- FRAGMENTED: only part of the BUSCO ID sequence has been found in the assembly.
- MISSING: the sequence of the BUSCO ID hasn’t been found in the assembly.
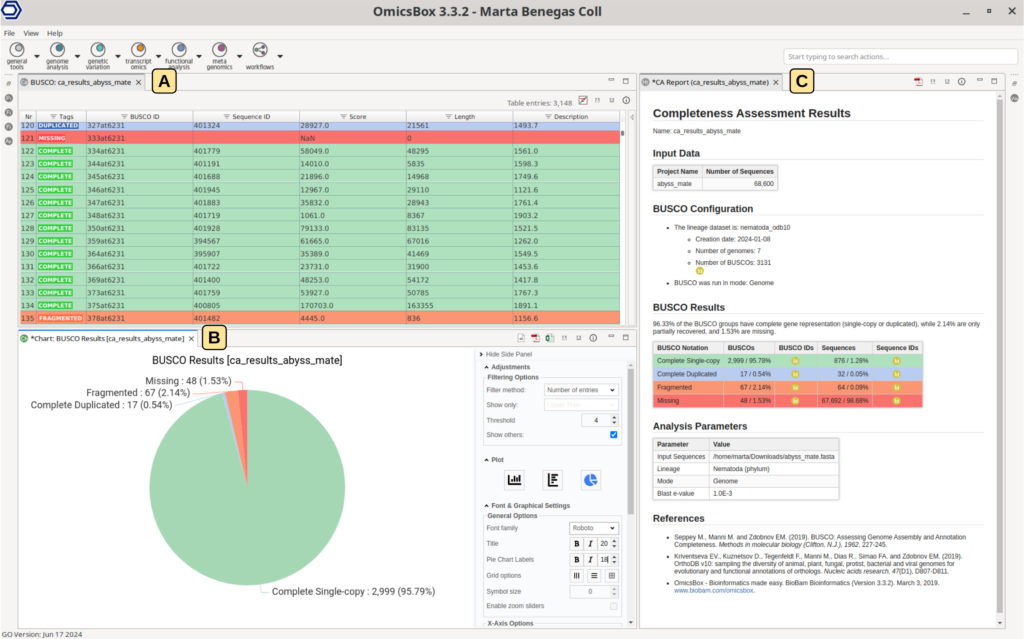
Moreover, this table shows the ID and the length of the sequence matching the BUSCO, as well as its alignment score.
A pie chart is also generated to easily assess the proportion of each tag at first glimpse (Figure 3-B). Finally, the report gives an overall summary of the analysis configuration and results (Figure 3-C).
Interpreting BUSCO results
A high percentage of complete BUSCOs is a good sign of a high-quality genome assembly. It means that core, conserved genes are present in their entirety, suggesting that the assembly is relatively accurate and has captured a large portion of the expected gene content.
A high percentage of duplicated BUSCOs can indicate issues with assembly, like over-assembly or contamination, leading to artificial duplications of regions. This is especially concerning if your organism is not expected to have many paralogs or gene duplications. Many duplicated BUSCOs could mean you’re dealing with unresolved heterozygosity (that is, alleles that have been detected and kept as different sequences) or that there are repetitive elements that haven’t properly collapsed during the assembly process.
Fragmented BUSCOs suggest that the assembly lacks continuity. This often happens when sequences are not long enough or are of low quality, leading to interruptions in the assembly of full-length genes. It can indicate that the genome is either under-assembled or that sequencing errors are preventing proper assembly. Many fragmented BUSCOs might mean that you need longer reads, better sequencing coverage, or improved assembly algorithms. It could also suggest that regions of the genome are difficult to assemble due to high complexity (e.g., repeat-rich regions).
Missing BUSCOs often point to gaps in the assembly where essential genes should be present but are absent. This could be due to low sequencing coverage, errors during the assembly process, or biological factors like gene loss in the species being studied (which would be less of a concern if it’s expected). If you have many missing BUSCOs, it’s a sign that your assembly is incomplete or that coverage in certain regions is insufficient. Missing BUSCOs can also suggest errors in genome annotation or sequencing bias, where some regions of the genome were underrepresented or missed entirely.
Key Points
- High complete, low duplicated/fragmented/missing: This is what you’re aiming for—a well-assembled genome.
- High duplicated BUSCOs: Investigate for over-assembly, contamination, or issues with heterozygosity.
- High fragmented BUSCOs: You might need better continuity, more data, or different assembly parameters to resolve these gene regions.
- High missing BUSCOs: Indicates incomplete assembly or insufficient data, suggesting that additional sequencing or reassembly may be necessary.
Conclusion
BUSCO is a valuable tool for identifying potential weaknesses or errors in genome assemblies, particularly those that may arise during the assembly process. When used in conjunction with QUAST, it forms a powerful pipeline for evaluating both the completeness and the structural integrity of de novo genome assemblies. QUAST provides detailed insights into assembly accuracy, while BUSCO focuses on biological completeness by assessing core conserved genes.
Together, these tools offer a comprehensive approach to genome quality assessment, allowing researchers to compare assemblies generated with different datasets, tools, and parameters. This combined methodology helps ensure that the resulting assemblies are of high quality and suitable for downstream analyses, ultimately empowering users to produce robust genome drafts that serve as reliable foundations for further study.
References
Seppey M., Manni M. and Zdobnov EM. (2019). BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods in molecular biology (Clifton, N.J.), 1962, 227-245.
Kriventseva EV., Kuznetsov D., Tegenfeldt F., Manni M., Dias R., Simao FA. and Zdobnov EM. (2019). OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial, and viral genomes for evolutionary and functional annotations of orthologs. Nucleic acids research, 47(D1), D807-D811.
OmicsBox – Bioinformatics Made Easy, BioBam Bioinformatics, September 6, 2024, OmicsBox by BioBam.
")
