Release OmicsBox version 1.2 (24th of October, 2019)
We are happy to announce the following updates for the transcriptomics module.
New features include Completeness Assessment and Predict Coding Regions.
More details can be found below as well as in the online user manual and Transcriptomics Module website.
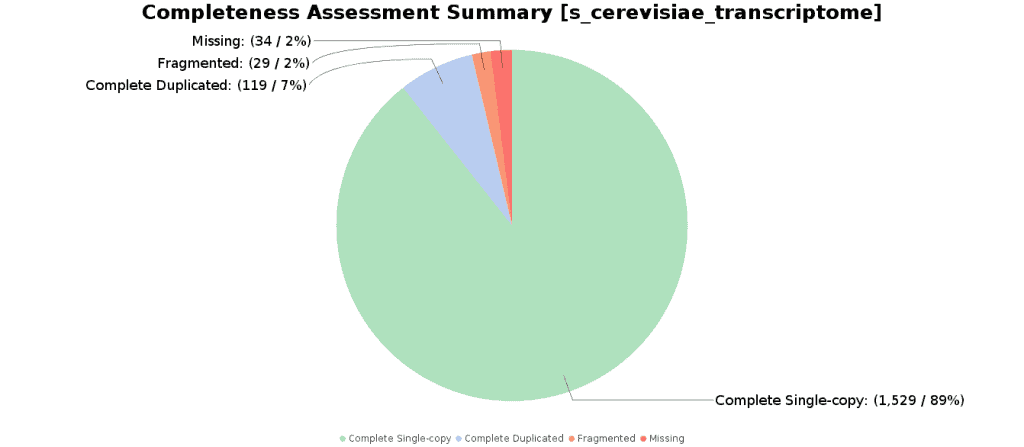
Completeness Assessment
The Completeness Assessment functionality provides quantitative measures for the assessment of transcriptome assembly completeness, based on evolutionarily-informed expectations of gene content from Benchmarking Universal Single-Copy Orthologs (BUSCO) selected from OrthoDB. The Benchmarking Universal Single-Copy Orthologs are ideal for such quantifications of completeness, as the expectations for these genes to be found in a genome/transcriptome in single-copy are evolutionarily strong.
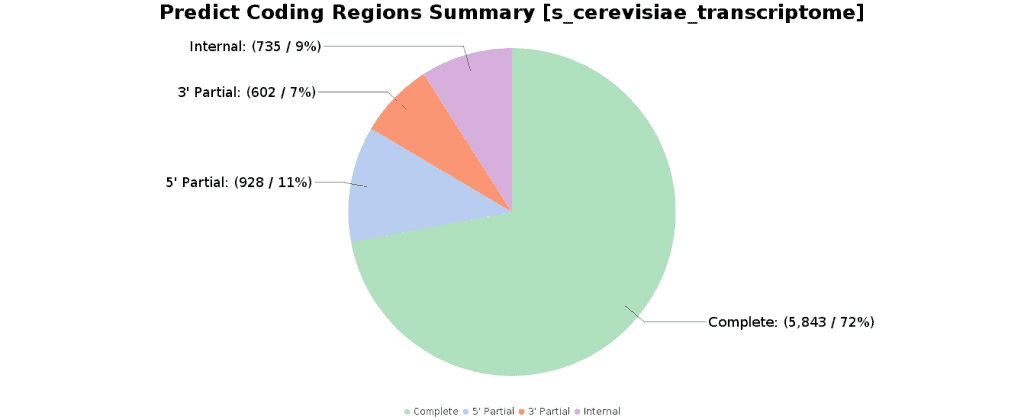
Predict Coding Regions
The Predict Coding Regions functionality detects candidate coding regions within transcript sequences, such as those generated by de novo RNA-Seq transcript assembly. It is based on TransDecoder, a pipeline that recognizes likely coding sequences based on the following criteria:
- A minimum length open reading frame (ORF) is found in a transcript sequence.
- A log-likelihood score is computed and it should be > 0.
- The above coding score is higher when the ORF is scored in the 1st reading frame as compared to scores in the other 2 forward reading frames.
- If a candidate ORF is found fully encapsulated by the coordinates of another candidate ORF, the longer one is reported. However, a single transcript can report multiple ORFs (allowing for operons, chimeras, etc).
- A Position-Specific Scoring Matrix (PSSM) is built, trained and used to refine the start codon prediction.
- The putative peptide has a match to a Pfam domain above the noise cut-off score (optional).
Minor Improvements
- New RNA-Seq Alignment Options:
- GFF is automatically converted to GTF to improve the performance.
- The 2-pass Mapping option allows a most sensitive novel junction discovery.
- Save spliced junctions in a tab-delimited format.
- Save unmapped and partially mapped reads in FASTQ format.
- New RNA-Seq de Novo Assembly Option:
- The Construct Super Transcripts option allows constructing “super transcripts” by collapsing unique and common sequence regions among splicing isoforms into a single linear sequence. Super transcripts provide a gene-like view of the transcriptional complexity of a gene.



