With improvements in accuracy and steadily decreasing costs, long-read sequencing technologies, such as the platforms provided by Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), have revolutionized the field of transcriptomics in recent years. Compared to short-read sequencing, which requires RNA fragmentation, long-read sequencing technologies can sequence RNA molecules in their entirety. This allows researchers to study the transcriptome at a transcript-, rather than merely a gene-resolution, shedding light on more complex alternative splicing events. To support researchers in analyzing such data, OmicsBox includes a variety of bioinformatic tools tailored to the analysis of long-read transcriptomics data.
The increasing adoption of such technologies in the community has been accompanied by a rich literature of bioinformatic tools to facilitate the analysis of the vast amounts of produced data. One significant challenge for the analysis of long-read RNA-sequencing data remains the identification of alternative RNA isoforms. While many tools exist to categorize transcripts based on comparing reads to reference genomes and transcriptomes (e.g. FLAIR and IsoQuant), such algorithms are of limited use to researchers interested in non-model species. For organisms where reference genomes and transcriptomes are not available or reliable, long-read RNA-sequencing data can still be characterized into RNA isoforms using the isON-pipeline of tools.
The isON-pipeline consists of the tools isONclust[1], isONcorrect[2], and isONform[3] and can process both ONT and PacBio data.
In the first step, isONclust, similar reads are clustered together. In the absence of a reference genome, this can be a challenging task, as different genes may be quite similar, or the same gene can create RNA isoforms with different exons.
The next step, isONcorrect, is only applied to ONT data. Here, the reads of each cluster can be compared to correct sequencing errors based on a consensus of similar reads.
Finally, isONform categorizes the clustered reads into transcript isoform sequences.
In OmicsBox, each of these steps can be easily customized to adjust the algorithms’ parameters for different use cases.
How to use the isON-pipeline in OmicsBox
1.Introduce Long-Read Data: Import the long-read data (as FASTQ). Pre-processing should already have been performed at this stage. Furthermore, due to excessive computational resource requirements and time constraints, short and long sequences are filtered out.
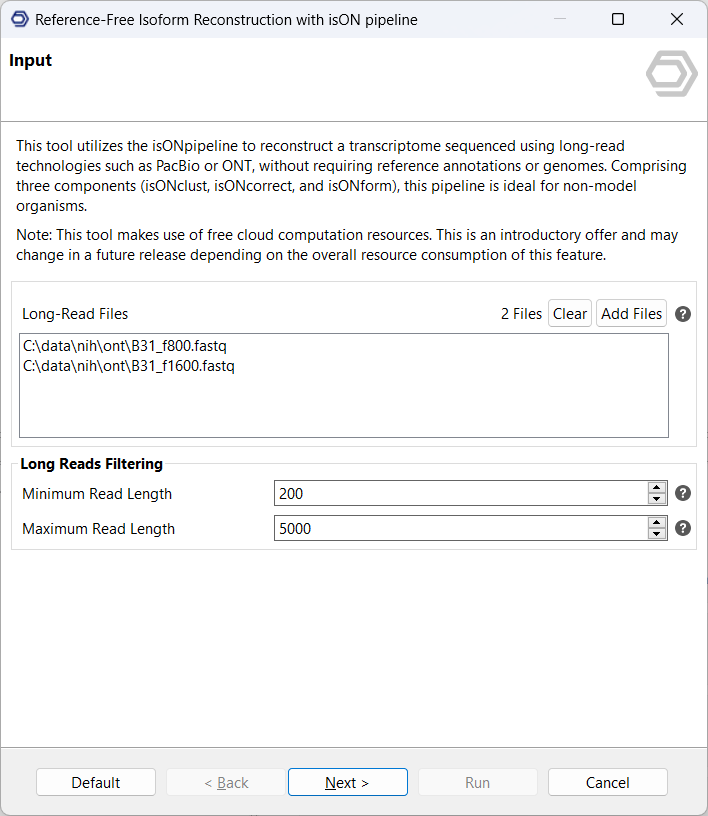
2.Configure the pipeline: Parameters of the individual steps can be adjusted. Most importantly, the data type (PacBio or ONT) has to be determined to choose the corresponding workflow design. Further information on the adjustable parameters is provided on the user manual page.
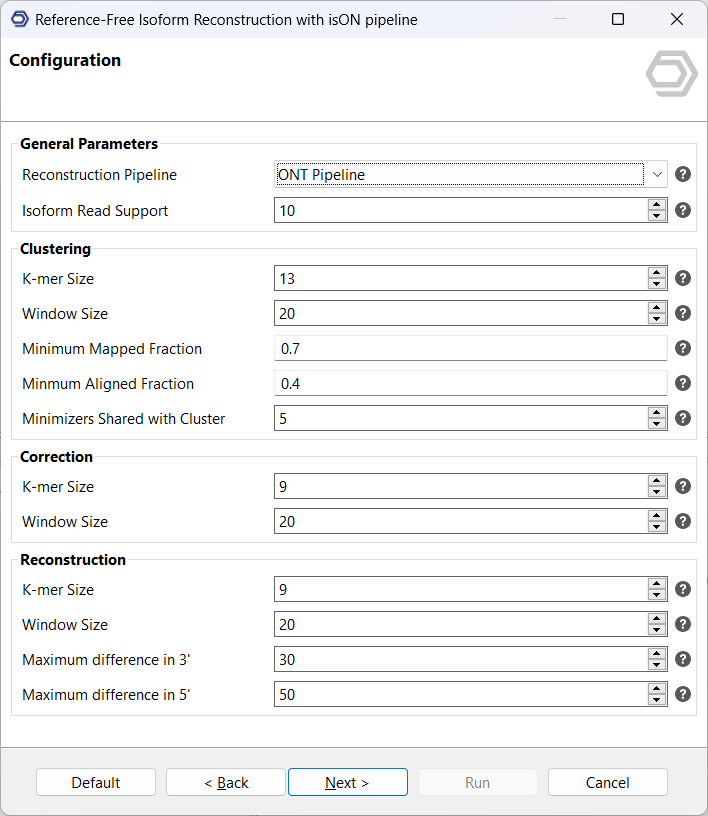
3.Configure output locations: As output files, a FASTA file with RNA isoform sequences is received, as well as a quantification file that reflects the number of supporting reads of each isoform, grouped by input files.
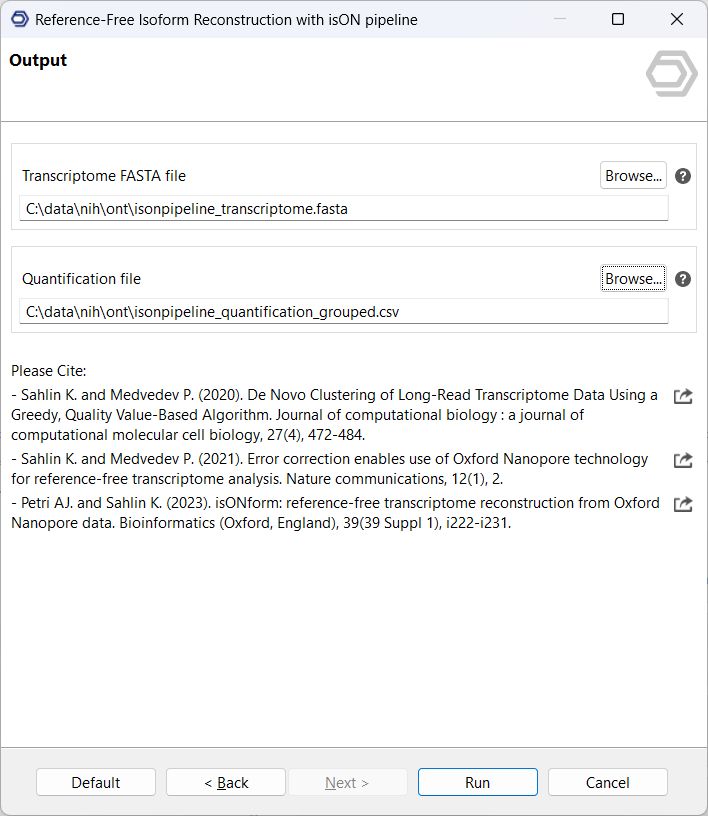
Results
Due to the lack of reference information, both the results, as well as downstream analyses are notably different between the isON-pipeline and other long-read RNA-sequencing transcript identification tools.
While assembled transcriptomes are usually presented in file formats such as GTF or GFF, these formats require an underlying reference genome. As a consequence, the transcript sequences identified by the isON-pipeline are provided as individual sequences in a FASTA format file.
In addition to the transcript sequences and their quantification, OmicsBox also provides customizable plots showcasing the distribution of the sequence lengths of discovered isoforms, as well as the distribution of the number of supporting reads of discovered isoforms.
Further downstream analyses may include conducting functional annotation of the transcript sequences using the Functional Analysis module.
The quantification of isoforms can also be used in a differential expression analysis. If the data stems from an experiment that includes different tissues, conditions, or time points, the differences in the expression of identified isoforms can be studied. Depending on the nature of the experiment and its design, this is can be achieved with edgeR, NOISeq, or maSigPro.
In summary, the isON-pipeline is a powerful set of tools that enables a new class of analysis. When available, the use of reference genomes and transcriptomes should still be preferred for the analysis of long-read RNA-sequencing data. However, thanks to the isON-pipeline, experiments on species where such reference information is unavailable or unreliable can still be analyzed. This potentially allows researchers to conduct novel experiments to discover new biological insights.
References
[1] Sahlin, Kristoffer, and Paul Medvedev. “De novo clustering of long-read transcriptome data using a greedy, quality value-based algorithm.” Journal of Computational Biology 27.4 (2020): 472-484. https://doi.org/10.1089/cmb.2019.0299
[2] Sahlin, Kristoffer, and Paul Medvedev. “Error correction enables use of Oxford Nanopore technology for reference-free transcriptome analysis.” Nature communications 12.1 (2021): 2. https://doi.org/10.1038/s41467-020-20340-8
[3] Petri, Alexander J., and Kristoffer Sahlin. “isONform: reference-free transcriptome reconstruction from Oxford Nanopore data.” Bioinformatics 39.Supplement_1 (2023): i222-i231. https://doi.org/10.1093/bioinformatics/btad264

