Transcriptome reconstruction is a challenging bioinformatic problem. The development of long-read sequencing technologies has made it easier to solve this issue thanks to the possibility of having transcripts entirely contained in a read. In addition, different algorithms have emerged to generate transcriptomes from long-read datasets, such as FLAIR and StringTie2. Both tools can be used for transcriptome reconstruction with long-read data. However, they employ distinct strategies to achieve it and they might be suitable for different applications.
The core difference is conceptual: while FLAIR tries to collapse long reads to generate isoforms, StringTie2 tries to assemble reads. Put another way, FLAIR focuses on reconstructing full-length isoforms, while StringTie2 reconstructs isoforms based on potential splice sites identified from the reads. In this analysis, we found out that FLAIR might be better at identifying annotated isoforms and accurately reconstructing the isoforms that are already known, while StringTie2 may be better at discovering unannotated transcripts.
Methods: StringTie2 and FLAIR
We compared FLAIR and StringTie2 in terms of precision and the number of transcripts identified. Precision refers to a tool’s ability to correctly identify true transcripts and is calculated by dividing the number of transcripts found by the tool by the total number of transcripts in a reference annotation.
We used datasets from 3 different species: Arabidopsis thaliana, Mus musculus, and Homo sapiens. On the whole, we conducted 9 analyses (three per species).
In the case of StringTie2, an original paper (Shumate et al., 2022) is the source of the metrics. In the case of FLAIR, we executed the following workflow using the Long Reads Toolset in OmicsBox:
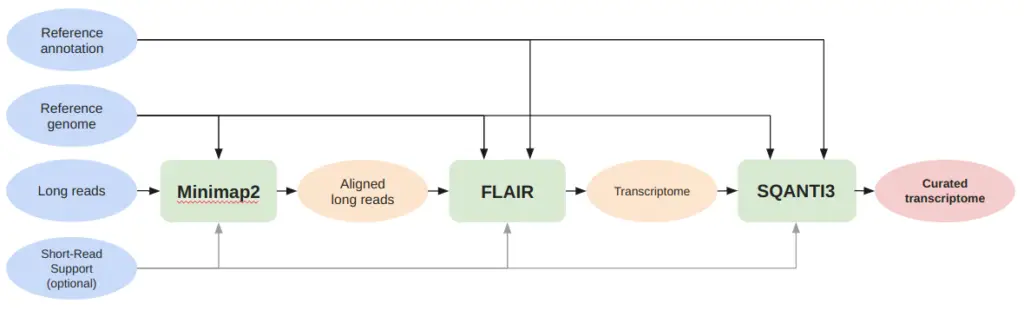
As shown in Figure 1, the pipeline begins with the use of Minimap2 to map long reads to the reference genome, utilizing specific parameters for the sequencing technology used for each dataset (ONT or PacBio).
Next, we employed FLAIR to generate a transcriptome for each long-read dataset, using a BAM file of the aligned reads. If a BAM file is not provided by the user, FLAIR uses Minimap2 to align the reads. However, for our pipeline, we utilize the BAM file generated earlier with specific parameters for the sequencing technology used. Finally, SQANTI3 is used to characterize the transcriptome and filter out unreliable transcripts, resulting in a curated transcriptome compared with the reference genome to determine the pipeline’s precision.
Once we obtained the curated transcriptome (the red circle in Figure 2), gffcompare is executed using the reference annotation to determine the precision and the number of transcripts found.
gffcompare -r reference_annotation.gtf curated_transcriptome.gtfDepending on the tool used, ‘Short-read supporting information’ encompasses different files. In the case of Minimap2 and FLAIR, we need to introduce BAM files of short reads (obtained also in OmicsBox using STAR). In the case of SQANTI3, the short reads are in FASTQ format.
Data Set
The previously described pipeline was applied to the following datasets in order to calculate the precision and the number of transcripts identified by the OmicsBox workflow:
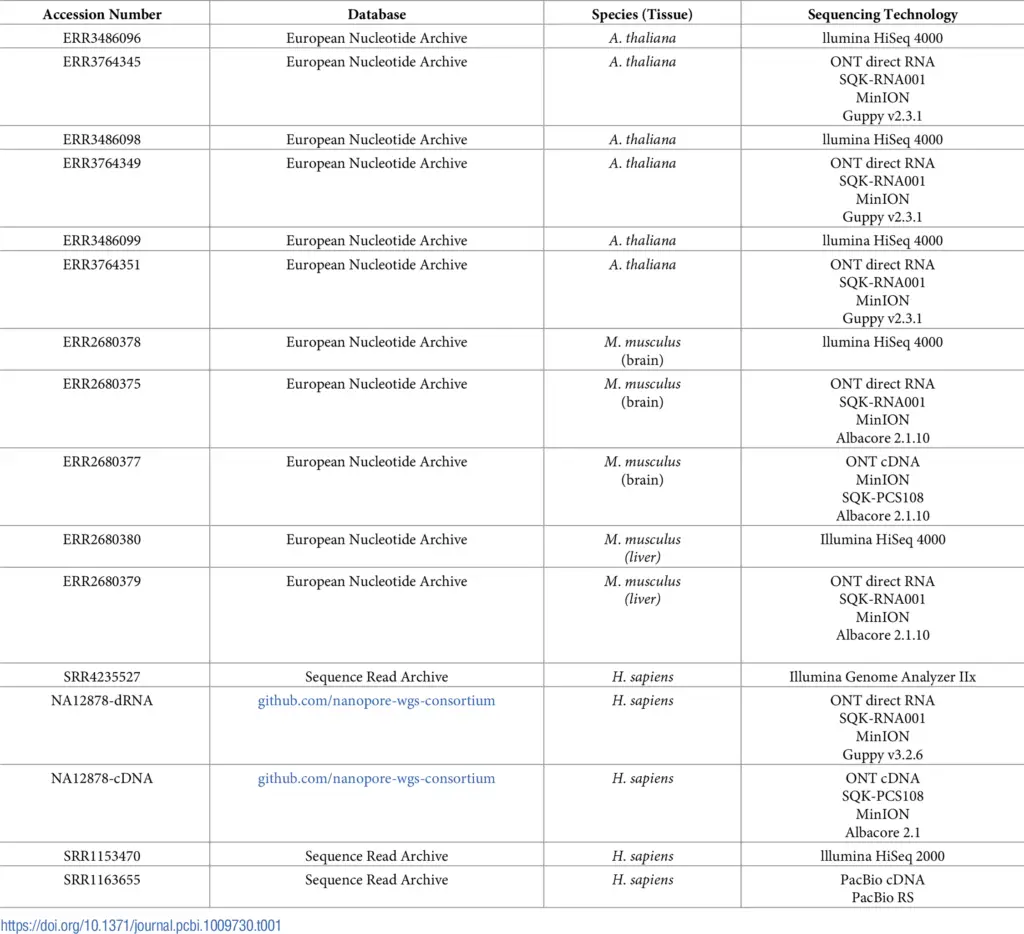
In total, we performed 3 analyses per species. Each long-read dataset was complemented by a short-read dataset in different steps in order to correct reads and splice junctions. The pairs of datasets used are
| Species | Datasets |
|---|---|
| A. thaliana | ERR3764345 and ERR3486096ERR3764349 and ERR3486098ERR3764351 and ERR3486099 |
| Mus musculus | ERR2680375 and ERR2680378ERR2680377 and ERR2680378ERR2680379 and ERR2680380 |
| Homo sapiens | NA12878-cDNA and SRR4235527NA12878-dDNA and SRR4235527SRR1163655 and SRR1153470 |
Comparison between StringTie2 and FLAIR
In the following chart, precision has been plotted against the number of transcripts for each dataset and tool.
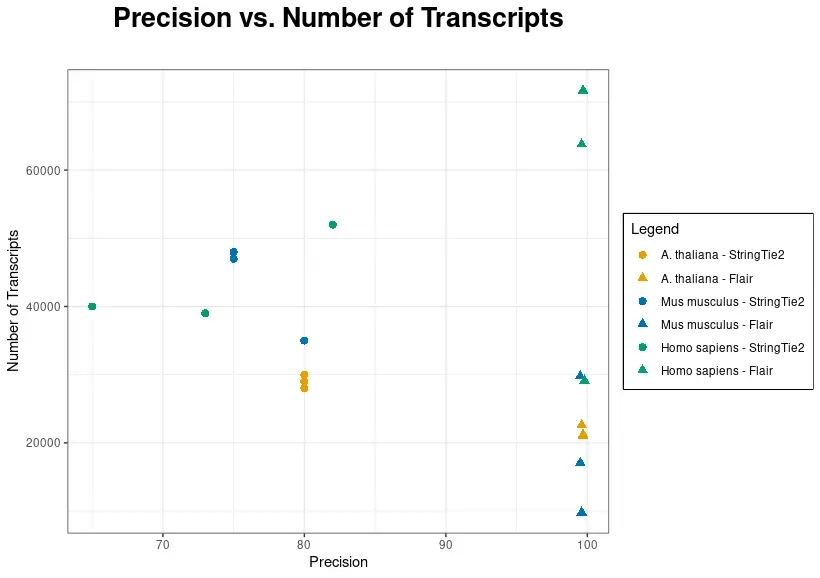
As shown in Figure 3, FLAIR precision is significantly higher. Nevertheless, the number of transcripts is generally higher when using StringTie2, although FLAIR recovers more transcripts than StringTie2 in two out of three datasets in Homo sapiens. Species with a reliable and well-established reference annotation may benefit from FLAIR, as it has the potential to identify a greater number of transcripts.
Conclusions
Results indicate that FLAIR might be the best option to have a clean transcriptome, without false positive transcripts, although some minor isoforms might not be present in the transcriptome. Moreover, FLAIR might overperform StringTie2 both in precision and the number of transcripts when a good reference annotation is provided. Nevertheless, in the case of building a new uncharacterized transcriptome, StringTie2 seems a better option.

Useful Links
- OmicsBox User Manual: Long-Read Characterization with SQANTI3
- OmicsBox User Manual: Long-Read Isoform Definition with FLAIR
References
- Shumate, A., Wong, B., Pertea, G., & Pertea, M. (2022). Improved transcriptome assembly using a hybrid of long and short reads with StringTie. PLoS computational biology, 18(6), e1009730.
- Tang, A. D., Soulette, C. M., van Baren, M. J., Hart, K., Hrabeta-Robinson, E., Wu, C. J., & Brooks, A. N. (2020). Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nature communications, 11(1), 1438.
- Tardaguila, M., De La Fuente, L., Marti, C., Pereira, C., Pardo-Palacios, F. J., Del Risco, H., … & Conesa, A. (2018). SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Research, 28(3), 396-411.
- Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 34(18), 3094-3100.
About the Author

With a biological and technological academic background, including a BSc in Biotechnology and an MSc in Bioinformatics, Enrique’s expertise lies in the areas of Long Reads and Genetic Variation.
")


