Functional genomics attempts to describe the functions of genes and proteins by making use of data derived from genomic and transcriptomic experiments. Some bioinformatics tools, like OmicsBox, offer biologists to get from raw NGS data through all steps up to the generation of biological insights for a species without any previous genome resources.
The bioinformatic process to perform a de novo transcriptomic analysis can be divided into 4 phases:
-
- Quality check and pre-processing of raw sequence data
- De novo transcript reconstruction and functional characterization
- Expression quantification and differential expression analysis
- Functional Enrichment Analysis
In this article, we will analyze the first point and we will explain how to perform NGS data preprocessing and quality control of raw sequence data.
FASTQ format and high-throughput sequencing
In the area of DNA and RNA sequencing, the FASTQ format has arisen as a standard file format for storing and sharing sequencing read data. In general, the FASTQ format provides an extension to the FASTA format, which allows for storing a numeric quality score associated with each nucleotide in a sequence. The FASTQ format has become widely used as a simple interchange file format. Unfortunately, the FASTQ format lacks a clear definition and it has several incompatible variants that depend on the sequencing platform.
On the other hand, high-throughput sequencing has accelerated genomic, transcriptomic, and metagenomic analyses. However, bioinformatic procedures are compromised by low-quality sequences, sequence artifacts, and sequence contamination, which can lead to misassembly and erroneous conclusions. To solve these problems is necessary to have better tools for quality control and preprocessing of all sequence datasets.
“The FASTQ format has arisen as a standard file format for storing and sharing sequencing read data”
What is a Quality Check and preprocessing?
A good quality control assessment and the corresponding preprocessing of raw sequence data from high throughput sequencing pipelines are fundamental for optimal downstream analysis.
Quality control for the raw reads involves the analysis of sequence quality, sequence length, GC content, the presence of adaptors, ambiguous bases, overrepresented k-mers, and duplicated reads in order to detect sequencing errors, PCR artifacts, or contaminations.
Acceptable duplication, k-mer, or GC content levels are experiments -and organism-specific, but these values should be homogeneous for samples in the same experiments. The preprocessing of data comprises a series of steps that involve handling low-quality base calls, trimming adapters from raw sequencing reads, and filtering of unwanted sequences.
Because these adapters are artificially introduced and are not part of the organism’s transcriptome, it is necessary to remove any remnants of them before attempting to do a downstream analysis.
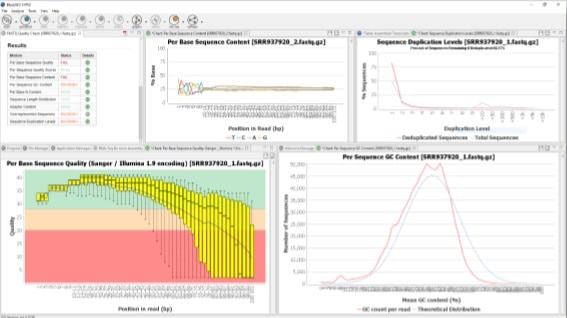
How to perform a Quality Check in OmicsBox
OmicsBox is a bioinformatics platform for high-quality functional annotation and analysis of genomic datasets and one of its functionalities is the FASTQ Quality Check. This tool provides an easy way to perform a quality control check on sequence data coming from high-throughput sequencing pipelines. The analysis is performed by nine modules that provide a quick overview of whether the data looks good and no problems or biases may affect downstream analysis. Besides, the results and evaluations are returned in the form of charts and tables. This functionality is based on the popular FastQC software.
To perform a FASTQ Quality Check it is necessary to have OmicsBox (you can download it here) and provide the following information:
-
- Raw Sequence Data: Select the FASTQ files containing the sequence data.
- Additional Adapter Sequences: This option allows to specify of a file that contains the list of adapter sequences that will be explicitly searched against the library.
- Additional Contaminant Sequences: This option allows you to specify a file that contains the list of contaminants to screen over-represented sequences against.
- Chart Read Length Binning: Enable grouping of bases for reads. If not, reports will show data for every base in the read.
Once finished, a new tab is opened containing simple composition statistics of each analyzed file. Each row corresponds to an input file, and columns show the following information: name, file type, encoding, total sequencing, Poor quality reads, Sequence Length, and %GC. Statistics and charts returned by each module can be displayed in an easy way.
For further information about how to perform a quality check in OmicsBox, please consult the user manual.
“OmicsBox is a bioinformatics platform for high-quality functional annotation and analysis of genomic datasets”
How to perform a preprocessing in OmicsBox
Preprocessing FASTQ files in OmicsBox consists of removing adapters and contamination sequences, trimming low-quality bases, and filtering short and low-quality reads. Before proceeding, it is advisable to carry out a quality control check of the sequencing data within OmicsBox (FASTQ Quality Check). In this way, problems and biases can be detected, which allows for better configure the preprocessing procedure.
The FASTQ Preprocessing tool uses the well-known preprocessing software Trimmomatic. Trimmomatic is a fast, multithreaded command line tool that can be used to trim and crop sequencing data as well as to remove adapters.
You can discover how to execute a FASTQ Preprocessing in OmicsBox in the user manual, but basically, this process is divided into the following phases:
-
- Adapter Removal: at this point, you have to enable the adapter removal step and then choose between using the default adapter sequences provided by Trimmomatic, or providing custom adapter sequences. In addition, other parameters can be set, like the number of allowed mismatches and the accuracy threshold.
- Trimming: here you can enable the trimming step and then select the strategy to perform the trimming step (Sliding Window Trimming, Adaptive Quality Trimming, Quality Trimming, and Length Trimming).
- Filtering: the penultimate step is Filtering by Quality and defining an Average Quality, and then filtering by Length and determining a Minimum Length.
As shown, OmicsBox allows in an intuitive way to easily perform a quality check and preprocessing raw sequence data. Do you need more information? Contact us.



